
FROM A POSSIBLE isolated outbreak in a wet market in Wuhan, China, Covid-19 has become a global pandemic affecting around five million people and killing more than three lakh worldwide. The causative organism is a virus that is so small that more than four lakh of its kind can fit on the tip of a needle. This new strain of coronavirus, named as novel coronavirus 2019, or 2019-nCoV, has an RNA (ribonucleic acid) genome of around 30,000 nucleotide bases and belongs to the betacoronavirus genus.
The first genome sequence of 2019-nCoV was isolated from a man employed at the Wuhan market. A consortium of researchers in China made the sequence available in the public domain; today, it serves as the reference point for understanding the virus and its evolution.
Like all organisms, coronavirus evolves through the accumulation of genetic mutations. Unlike the influenza viruses that cause the common flu, the 2019-nCoV mutates at a much slower pace. It is estimated that the virus accumulates one mutation approximately every 15 days. As the virus replicates and transmits, mutations get accumulated in its genome, thus forming different evolutionary groups or ‘clades’.
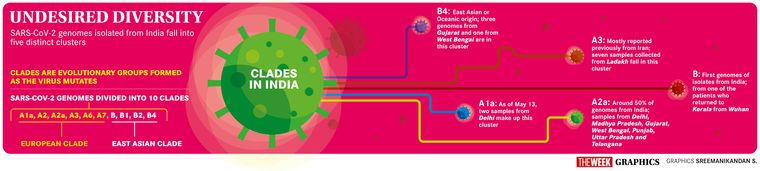
Sequencing the genome can provide a view of the genetic mutations in a particular strain and how it compares with the rest of the strains worldwide. Based on information from GISAID, a database hosted by the German government to share genomic data, the research network Nextstrain has broadly divided the 2019-nCoV genomes into 10 clades: A1a, A2, A2a, A3, A6, A7, B, B1, B2 and B4. The ‘A’ supergroup, or A superclade, are known as the ‘European clade’, since the sequences falling under this type originated in European nations. The B superclade is called the ‘East Asian clade’, also based on its origins.
The sequence of the viral genome thus provides researchers with an opportunity to understand how the virus evolves, and more importantly, how it spread across the world. The initial genomes of 2019-nCoV from India were obtained from one of the patients who had travelled from Wuhan to Kerala. The genomes were sequenced by the National Institute of Virology in Pune. Till date, over 200 2019-nCoV genomes from India have been deposited in public databases globally. These include isolates from government agencies such as the National Institute of Virology, the National Centre for Disease Control, the National Institute of Biomedical Genomics, and the National Institute of Mental Health and Neurosciences. The majority of genomes have been made available through a collaborative effort between the National Centre for Disease Control and the CSIR Institute of Genomics and Integrative Biology. The Gujarat Biotechnology Research Centre, a state-sponsored research organisation, has deposited more than 100 genome sequences of 2019-nCoV isolates collected from across Gujarat.
The genomes can be compared based on the genetic mutations they have. This comparison can help map out a visual construction of what is known as a phylogenetic tree—a family tree of the virus that depicts how the different genome sequences are related to each other. Clades can thus be identified on the tree as a cluster that shares a common ancestor and descends from the same branch.
Phylogenetic analysis has shown that the Indian coronavirus isolates largely cluster into five clades—A1a, A2a, A3, B and B4. Most of the Indian genomes fall in the A superclade, with a majority encompassing A2a and A1a clades and a few in A3 clade. A2a is a globally predominant clade. The genomes in A3 clade, which was mostly reported in Iran earlier, are from isolates collected from Ladakh.
The initial genomes from Kerala fell into the B clade, and are from individuals who had travelled from Wuhan. The recent addition of B4 clade to the Indian cluster was largely through the sequencing efforts of Gujarat Biotechnology Research Centre and the National Institute of Biomedical Genomics in West Bengal. The B4 clade is a sub-type of the superclade B with potential origins from either East Asia or Oceania. Sequences that belong to the B4 clade harbour two distinguishing mutations in their genomes. The first mutation, L84S, in the gene ORF8 is common among all clades of the B superclade. The other mutation is S202N in the gene that encodes the nucleocapsid protein of the virus. In the phylogenetic tree of genomes from India, three genomes from Gujarat and one from West Bengal fall under this cluster.
A number of people suggest that the clades have a difference in severity, but such claims have not been proven. Though some clades are predominantly prevalent in some locations than in others, there is insufficient data to draw conclusions about the differences in virulence and clinical outcomes of these clades. More genomes and systematic tagging of clinical information for the genomes would significantly improve our understanding in this direction.
The Covid-19 pandemic has highlighted the need to share genomic data on a global scale. Researchers from more than 70 countries have made over 30,000 coronavirus genomes publicly available—one of the best examples of open data initiatives shaping up across the globe. It also highlights the renewed interest in open source movements for developing better diagnostics and novel therapeutics, something that has been rightly emphasised by World Health Organization director-general Tedros Adhanom.
—The authors are researchers at the CSIR Institute of Genomics and Integrative Biology (CSIR-IGIB), Delhi.